
Advances in Animal and Veterinary Sciences


Short Communication
Advances in Animal and Veterinary Sciences 2 (2S): 27 – 30Special Issue–2 (2014) (Advances in Diagnosis and Control of Infectious Diseases of Animals)
Sequencing of Envelop Protein P32 Gene of Vaccine Strain of Sheeppox Virus
Kanisht Batra1, Narender Singh Maan2, Aman Kumar1, Arnab Ghosh1, Sunayna1, Sushila Maan1*,
- Department of Animal Biotechnology, College of Veterinary Sciences, Lala Lajpat Rai University of Veterinary and Animal Sciences, Hisar, 125 004, Haryana, India
- Department of Animal Nutrition, Resource faculty Department of Animal Biotechnology, College of Veterinary Sciences, Lala Lajpat Rai University of Veterinary and Animal Sciences, Hisar, 125 004, Haryana, India
*Corresponding author:[email protected]
ARTICLE CITATION:
Batra K, Maan NS, Kumar A, Ghosh A, Sunayna, Maan S (2014). Sequencing of envelop protein P32 gene of vaccine strain of sheep pox virus Adv. Anim. Vet. Sci. 2 (2S): 27 – 30.
Received: 2014–03–08, Revised: 2014–04–12, Accepted: 2014–04–13
The electronic version of this article is the complete one and can be found online at
(
http://dx.doi.org/10.14737/journal.aavs/2014/2.2s.27.30
)
which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
ABSTRACT
Capripox viruses are the etiological agents of economically devastating poxviral diseases in cattle, sheep and goats and are endemic in India. The aim of this study was to genetically characterize a vaccine strain of sheeppox virus (Rumanian Fanar strain) by determining the sequence of P32 gene and its comparison publically available sequences.
Sheeppox (SP) is an infectious disease of small ruminants responsible for high economic losses to developing country such as India. There have been several reports of pox in sheep from different parts of India (Sharma et al., 1986; Rao et al., 2000). Sheeppox virus belongs to Genus Capripoxvirus (CPV) of family Poxviridae. The genus Capripoxvirus comprises three members namely, sheeppox virus (SPV), goatpox virus (GPV) and lumpy skin disease virus (LSDV) affecting sheep, goats and cattle, respectively. Sheepox disease is associated with significant production losses because of reduced milk yield, decreased weight gain, increased abortion rates in pregnant animals, damage to wool and hides, increased susceptibility to pneumonia and other secondary infections. Disease incidence is high in pure and crossbred lambs and hoggets but morbidity is more in lambs, yearlings and immunologically naive sheep. The disease is prevalent throughout India but there are more reported regular outbreaks in Karnataka, Jammu, Maharastra and Tamilnadu states (Bhanuprakash et al., 2006). There are various predisposing factors of the disease, which can be host related, virus agent related and environment related. Host related factors include: age, sex, breed, nutritional and immunological status of the animal, whereas agent related factors consist of strain, virulence, and pathogenicity. Poor management and feed scarcity are environment factors that directly affect occurrence of the disease.
There are various methods such as agar gel immune–precipitation test (AGPT), counter–immunoelectrophoresis (CIEP), enzyme linked immunosorbent assay (ELISA) and virus neutralization tests (VNT) had been described for virus identification but not widely acceptable due to lack of sensitivity, specificity and presence of cross reactivity (Bhambani, 1963; Uppal, 1967). However, nucleic acid based techniques such as PCR, cloning and sequencing are more reliable, sensitive and specific techniques. In this report the authors describes amplification, cloning and sequencing of P32 gene of vaccine strain (Rumanian Fanar strain) of SPV.
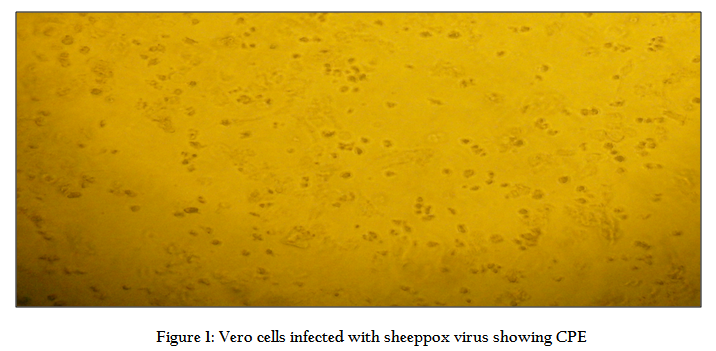
Lyophilized Indian vaccine strain (Rumanian Fanar) was procured from Haryana Veterinary Vaccine Institute (HVVI), Hisar. The vaccine virus was reconstituted in 1 mL PBS and was used to infect Vero cells (derived from African green monkey kidney) as described by Amal, (2008) and Mangana–Vougiouka et al. (1999). Infected culture was grown in MEM supplemented with L–glutamine (0.03%) and antibiotics (100 µg streptomycin and 100 IU penicillin/100 mL). After 5 days post–inoculation, infected cells showing >80% cytopathetic effect (CPE) were harvested (Figure 1). CPE appears as granulation of cells followed by rounding, ballooning and aggregation along with detachment of cell sheath during successive 4–5 days of post inoculation. Infected cultures were pelleted down by centrifugation at 2,500 rpm for 15 min. The pellet was then resuspended in 250 µL of phosphate buffer saline (PBS) and used for isolation of viral DNA.

Pellet suspended in PBS was treated with 500 ul lysis buffer (prepared by adding 3.3 mL of 3M sodium acetate (pH 5.2) and 5 mL of 10% SDS with addition of distilled water to make final volume as 100 mL) (Sambrook et al., 1989) 0.1 volume of 2M Sodium acetate, 0.1 volume 10% SDS, 20 µL proteinase K (20 mg/mL) and incubated at 55OC for one hour. Clear solution obtained after incubation was subjected to treatment with equal volume of phenol: chloroform: isoamyl alcohol (PCI: 25:24:1). Contents were mixed and centrifuged at 12,000 rpm for 15 min. Supernatant was further treated with CI (24:1) to remove traces of phenol. After centrifugation, supernatant was precipitated overnight using two volumes of absolute ethanol and 3M sodium acetate (pH 5.2). DNA precipitated after incubation was washed with 70% alcohol to remove excess salt and air dried pellet was suspended in 30 µL of nuclease free water. The purified DNA was quantitated using Qubit® dsDNA HS (High Sensitivity) Assay Kit for use with Qubit® 2.0 Fluorometer (Invitrogen).
Three pairs of primers were designed to amplify the complete P32 gene of SPV using Bioedit sequence alignment software. Flanking primer pair (SPV–P32/FF and SPV–P32/FR) was used for amplification of full length P32 gene of expected size 1145 bp (Figure 2). Another pair of primer consisted of two terminal primers (SPV–P32/TF and SPV–P32/TR) to amplify coding region of P32 gene of expected size 972 bp. A pair of internal primer was also designed to use in combination with terminal primers to give an amplicon of 578 bps and 581 bps (Table 1). PCR conditions were standardized with the DNA isolated from vaccine strain with appropriate positive and negative controls. PCR was performed in 25 µL reaction containing 5 µL of template DNA (400 ng) in high fidelity Fusion Taq mastermix (2x concentration) with GC buffer (NEB). Primer concentration of 0.4 µM of each forward and reverse primer was used. Cyclic conditions for PCR used were: Initial denaturation at 94OC for 3 min, and 35 cycles of denaturation at 94OC for 30 sec, annealing at 48OC for 45 sec, elongation at 72OC for 45 sec followed by final elongation at 72OC for 10 min. An aliquot of 2 µL were analyzed in 1% agarose gel containing ethidium bromide. P32 gene amplicons of 578 and 581 bps were obtained when visualized under gel documentation system (Bio–Rad) (Figure 3).
Figure 2: Amplification of full length P32 gene of sheeppox virus using flanking primer pair (pair 1; Table 1)
Figure 3: Amplification of P32 gene of sheeppox virus using gene specific primer pairs 3 and 4 (Table 1)
The full length P32 envelope gene after purification using gel extraction kit (Qiagen) was prepared for cloning in a CloneJET™ PCR Cloning Kit (Thermo Scientific K1230). Purified amplicons of sizes 578 and 581 were used for ligation in cloning vector. Ligation mixture of 20 µL was prepared containing 10 µL of 2 x reaction buffers, 1 µL p JET 1.2 /Blunt cloning vector (50 ng/ µL), 1 µL of T4 DNA ligase and 150 ng of purified PCR product. The optimal insert / vector ratio for ligation was 3:1. Ligation mixture was incubated at 22OC for 5 min and used for transformation.
Competent cells were prepared from GM2163 E. coli strain after subculturing from overnight grown bacteria using 0.6 volume of 4:1 MgCl2 and CaCl2. After treatment cells were centrifuged at 5000 rpm for 10 min and pellet was resuspended in 500 µL of CaCl2. Transformation was done by adding ligation mixture to competent cells, subjecting it to heat shock at 42OC for 2 min, followed by snap chilling on ice and incubation at 37OC for 1 hr. Screening and selection of recombinant clones was done on basis of expression of a lethal gene on plates containing ampicillin. Positive colonies are first confirmed by colony touch PCR using gene specific primer pairs 3 and 4 as listed in Table 1 and shown in Figure 3. Plasmids were isolated using plasmid isolation kit from Invitrogen as per given procedure.

Figure 4: Unrooted neighbor–joining phylogenetic tree drawn from complete sequences (972 bp) of P32 gene of sheeppox virus; The tree was constructed using a p–distance algorithm and pairwise deletion parameters;
Recombinant plasmids were sequenced using BigDye terminator v3.1 cycle Sequencing kit from Applied Biosystems on a ABI 3130XL genetic analyser. The sequence data generated was assembled using SeqMan programme of the Lasergene software ver 5.0. The contig sequence obtained was submitted to Genbank under accession number KJ679574 and aligned with the available sequences in the Genbank using MEGA 5.0 programme. The sequence analysis of envelope protein (P32) gene of RF strain obtained from HVVI, Hisar showed that the gene is highly conserved with >99% nucleotide sequence identity among global strains. The P32 gene sequence of vaccine strain studied here showed 99.9% nucleotide identity with other SPV strains from India (Srinagar and Makhdoom) and China (Accession numbers JN596272 and JN596273) as calculated using MEGA 5.0 programme. Minimum nucleotide identity is 99.1% with AV40 strain from China (Accession number HQ607368).
In conclusion, the sequence generated from the vaccine strain of sheepox virus is a valuable addition to global sequence database that can be helpful in designing appropriate highly sensitive and specific tests such as probe based assays or dye based assays.
AUTHOR’S CONTRIBUTIONS
SM, NSM, AK: conception and design of experiments, drafting the manuscript, provided reagents and facilities. Sunayna, MS, AG, KB, AK: acquisition and analysis of data. All authors have read and approved the manuscript.
ACKNOWLEDGEMENTS
This work was funded by Rastriya Krishi Vikas Yojna scheme no. 4011/C (g) ABT–4–0A. The authors are thankful to Dr. Ravindra Sharma, Director of Research for providing all required facilities. The assistance provided by non–teaching staff particularly Mr. Chandan Singh is also highly appreciated.
COMPETING INTERESTS
The authors declared that they have no competing interests.
REFERENCES
Amal MAR, Shahein MA, Hoda AEM (2008). Electrophoretic characterization of sheep pox virus, goat pox virus and lumpy skin disease virus. Egypt J Comp Path & Clinic Path 21(1):15–30.
Bhambani BD, Murty DK (1963). An immuno–diffusion test for laboratory diagnosis of sheeppox and goatpox. J Comp Pathol 73:349–358.
http://dx.doi.org/10.1016/S0368-1742(63)80037-5
Bhanuprakash V, Indrani BK, Hosamani M, Singh RK (2006). The current status of sheep pox disease. Comp Immunol Microb 29:27–60.
http://dx.doi.org/10.1016/j.cimid.2005.12.001
PMid:16458357
Mangana–Vougiouka O, Martoulatos P, Koptopoulos G, Nomikou K, Bakandritsos N, Papadopoulos O (1999). Sheeppox virus identification by PCR in cell cultures. J Virol Methods 77:75–79.
http://dx.doi.org/10.1016/S0166-0934(98)00138-4
Sambrook J, Fritsch EF, Maniatis T (1989). Molecular cloning: A Laboratory Manual. 2nd ed. New York: Cold Spring Harbor Laboratory Press.
Sharma MM, Uppal PK, Lonkar PS, Mathur PB (1986). Epidemiology of a sheep pox outbreak in mutton and fine wool type of sheep at an organised farm. Indian J Anim Sci 56(12):1183–1186.
Rao TVS, Bandyopadhyay SK (2000). A comprehensive review of goat pox and sheep pox and their diagnosis. Anim Health Res Rev 1(2)127–136.
http://dx.doi.org/10.1017/S1466252300000116
PMid:11708598
Uppal PK and Nilakantan PR (1967). Serological reactions in sheeppox– II. Agar gel diffusi

