
Advances in Animal and Veterinary Sciences


Research Article

Optimization of Recombinant Glycoprotein D (gD) based Indirect ELISA for Detection of Antibodies against Bovine Herpesvirus-1
Barkha Ratta1, Swagatika Priyadarsini1, Nikhil K. Channabasappa1, Pashupathi Mani1, Meeta Saxena1, R Saravanan2, Ajay Kumar1*
1Division of Biochemistry, Indian Veterinary Research Institute, Izatnagar, Bareilly (UP), 243122, India; 2Immunology Section, Indian Veterinary Research Institute, Izatnagar, Bareilly (UP), 243122, India.
Abstract | Bovine herpesvirus 1 (BoHV-1) affects mainly large ruminants (cattle and buffalo), resulting in a substantial negative impact on the industry. The virus causes respiratory symptoms, abortion and a decrease in milk production. Among envelope proteins of the virus, the glycoprotein D (gD) is considered a dominant immunogenic. We expressed the immunogenic portion of gD in the prokaryotic system (E. cloni 10G) and did evaluate its potential as a diagnostic antigen for indirect ELISA. Using the Ni-HRP probe, the expressed recombinant gD protein (rgD, 45kDa) was characterized by SDS-PAGE and western blotting. The immunogenic reactivity of red was optimized for indirect ELISA using known true positive and negative sera (antibodies) of the virus. Sera (n=158) were screened and, with a cut-off value of 0.238, relative diagnostic sensitivity (69%) and specificity (52.3%) of in-house developed indirect ELISA was determined. With these preliminary results, the study suggests the diagnostic potential of recombinant gD protein for serodiagnosis of BoHV-1 infection at a large scale.
Keywords | Indirect ELISA, Bovine herpesvirus-1, Glycoprotein D, Antibodies, Recombinant
Received | February 19, 2020; Accepted | June 21, 2020; Published | July 18, 2020
*Correspondence | Ajay Kumar, Division of Biochemistry, Indian Veterinary Research Institute, Izatnagar, Bareilly (UP), 243122-India; Email: [email protected]
Citation | Ratta B, Priyadarsini S, Channabasappa NK, Mani P, Saxena M, Saravanan R, Kumar A (2020). Optimization of recombinant glycoprotein D (gD) based indirect ELISA for detection of antibodies against bovine herpesvirus-1. Adv. Anim. Vet. Sci. 8(8): 853-860.
DOI | http://dx.doi.org/10.17582/journal.aavs/2020/8.8.853.860
ISSN (Online) | 2307-8316; ISSN (Print) | 2309-3331
Copyright © 2020 Ratta et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
INTRODUCTION
Bovine herpesvirus-1 (BoHV-1) primarily infects cattle and buffaloes and causes infectious bovine rhinotracheitis (IBR) and infectious pustular vulvovaginitis (IPV) along with other complications such as conjunctivitis, abortion, encephalitis, enteritis, and generalized disease in newborn (Muylkens et al., 2007). The BoHV-1 is a member of the subfamily Alphaherpesvirinae which includes Human herpesvirus-1 (HHV-1), a prototype virus for Alphaherpesvirinae (Misra et al., 1981; Mayank et al., 2018). One of the hallmarks of Herpesviruses (e.g., BoHV-1) is their ability to become latent in the host. soon after infection. and, after reactivation, the affected animal shed virus through nasal secretions and semen (Jones et al., 2007).
The IBR and IPV are among the endemic diseases of cattle in India as a result of crossbreeding. The virus was isolated and reported for the first time by Mehrotra et al. (1976) from crossbred calves at an organized cattle herd in Uttar Pradesh, who had a history of keratoconjunctivitis. Since then, the disease has been reported in most of the states of India.
The BoHV-1 has a 140 kb linear double-stranded DNA genome (Roizman, 1996) that encodes ten glycoproteins. Among them, glycoproteins gB, gC, and gD are highly immunogenic and, therefore, make them suitable candidate antigen for the development of immunodiagnostic assays. Currently, the disease is diagnosed by virus isolation, PCR, and serology (Gupta et al., 2006; Madbouly et al., 2008). Serum neutralization (SN) test is often used; however, it lacks sensitivity, time-consuming, and laborious (Balamurugan et al., 2010). For PCR reaction, aerosol contamination is much frequent, and an occurrence of false-positive results is not unusual. Consequent to these limitations of SNT and PCR, ELISAs could be potential alternates for antibody detection, which are proved to be fast, sensitive, and relatively inexpensive (Collins et al., 1985; House et al., 1971). Other than this, allowing the use of purified recombinant viral envelope proteins as antigenic substrate, ELISA can easily be applied on a large scale screening of test samples. These recombinant viral envelope proteins can be produced in large quantities without handling live viruses and, therefore, can eliminate the presence of host cellular proteins in the test and subsequent reduction of false-positive reactions (Balamurugan et al., 2010). With this background, we expressed recombinant BoHV-1 gD (rgD) in the prokaryotic system and used as diagnostic antigen in the development of a simple, rapid and reliable indirect ELISA for detection of antibodies against BoHV-1 infection.
MATERIALS AND METHODS
Virus and Cell culture
An Indian isolate of BoHV-1 (BoHV1/IBR216II/1976/India, not sequenced, only isolated) was procured from Immunohistochemistry Laboratory, Division of Biochemistry, IVRI, Izatnagar. BoHV-1 was propagated in Madin Darby Bovine Kidney (MDBK) cells. The cells were procured from NCCS (National Centre for Cell Science), Pune, India and maintained in 10 % DMEM media (GE Healthcare HyClone, USA) at 37℃ and 5% CO2 in a humidified CO2 incubator.
Amplification of BoHV-1 gD gene
Viral genomic DNA was isolated using PureLinkTMViral RNA/ DNA Mini kit (Invitrogen, Thermo Fisher Scientific) as per manufacturer’s instructions. The extracted DNA was amplified for gD gene using gene specific forward: SUMO/gD/F (5’AACAGATTGGAGGTGCCGTGCTGGGCGCG CTGCTCGCC3’) and reverse primer: SUMO/gD/R(5’GTGGCGGCCGCTCTATTAGCGGGCCTCGTCGTCGCC GGGTGGCG3’. Primers were designed from the NCBI sequence (NC_001847.1) as per the criteria mentioned in the kit (Expresso® Rhamnose SUMO Cloning and Expression System, Lucigen, SIGMA). The PCR cycling conditions were: 94°C for 5 min; followed by 35 cycles of 94°C for 45sec, 62°C for 45 sec and 72°C for 1 min and a final extension of 72°C for 10 min. The amplified products were analyzed in 1% agarose gel and documented.
Construction of expression cassette in pRhamTM N-His SUMO Kan vector
The amplicon was purified from the agarose gel using GeneJET Gel Extraction Kit (Thermo Scientific, USA) and cloned in pRhamTM N-His SUMO Kan vector. The insert (amplified gD gene) was co-transformed with the vector into chemically competent E. cloni 10G cells. Approximately, 100ng of insert mixed with 25ng (2µl) of vector was transformed into chemically competent E. cloni 10G cells (Lucigen, SIGMA, USA) by heat shock method. The recombinant transformed cells were plated on Luria Bertani (LB) agar containing kanamycin (50µg/ml), for the selection of recombinant clones. The colonies were screened by colony PCR, restriction enzyme (RE) analysis. They were further confirmed by DNA sequencing (DNA Sequencing Facility, Department of Biochemistry, Delhi University, India) for the identification of the cloned gene products.
Expression of recombinant gD protein
The positive colonies were inoculated in five tubes of LB broth (5 ml each) at 37 °C. Once culture reached mid-log phase (OD600 of 0.4–0.5), it was induced for expression of the cloned gene products. To optimize the induction time for expression of cloned gene products (rgD protein), uninduced culture were also collected subsequently. Two cultures were induced with 0.15% and 0.2% Rhamnose and incubated at 32 °C. The other two were induced with 0.15% and 0.2% Rhamnose and incubated at 37 °C for expression of recombinant gD protein. All the tubes were kept at 200 rpm in a shaker incubator. Cultures were collected at different time intervals of 2, 4, 6 and 8 hrs and pelleted, followed by cell lysis through boiling. The tubes were snap-chilled, thawed to room temperature, centrifuged and the supernatant was loaded into SDS-PAGE gel. Confirmation of the expressed protein was done by western blot using the Ni-HRP probe.
Finally, the recombinant protein expression was optimized to 0.15% Rhamnose at 32 °C for 8hrs.
Purification of expressed recombinant gD protein
Harvested 100ml of induced culture pellet at 8hrs post induction was suspended in 10ml of 1X binding buffer (0.5M NaCl, 20mM Tris base, 5mM Imidazole, 1mM PMSF,1mg/ml Lysozyme, pH 7.9) and mixed by pipetting in a 50ml tube and freeze thawed thrice. The suspension was sonicated for ten bursts with 15sec pulse and centrifuged at 3500g, 4oC for 30min. The pellet was resuspended again in 1X binding buffer carrying 6 M urea and centrifuged at 3500 g, 4oC for 30min. The supernatant was collected for affinity purification. Initially the Ni-NTA (Nickel- nitrilotriacetic acid) affinity chromatography column (MERCK, USA) was equilibrated thrice with 5ml of equilibration buffer (6M urea, 50mM Na2HPO4, 0.5M NaCl and 5mM imidazole, pH 8.0). The supernatant (isolated recombinant gD protein from bulk culture), was loaded on to the Ni-NTA column and the flow through was collected in a 15 ml tube and column was washed thrice with 5 ml of wash buffer (6M Urea, 50mM Na2HPO4, 0.5M NaCl and 20mM imidazole, pH8.0). After washing, the protein was eluted in 1ml fractions in elution buffer (6M Urea, 20mM Tris-HCl, 100mM NaCl and 400mM imidazole, pH 7.5). The eluates were collected and stored at -200C until further use.
Dialysis of the eluted protein was performed in dialysis buffer containing decreasing concentration of urea (6M, 4M, 2M, 1M) for refolding or renaturation of purified denatured protein. The final concentration of purified BoHV-1 rgD protein was estimated by Lowry protein estimation method (Lowry et al., 1951) and the rgD with protease inhibitors was stored in -80°C in aliquots.
The dialyzed protein was resolved by SDS-PAGE and further confirmed by Western Blotting. The resolved bands were transferred on to a polyvinylidine difluoride (PVDF) membrane (MDI, India) and the membrane was blocked with 3% BSA (Bovine serum albumin, SIGMA-Aldrich) in PBS-Tween 20 buffer (pH7.2) overnight at 4°C. The blot was incubated with Ni-HRP Probe and incubated for one hour followed by washing of membrane and development of blot with Diaminobenzidine (DAB) as a chromogen and documented.
Standardization of indirect ELISA
For establishing the optimal concentration of antigen and the serum dilution, different concentrations of purified rgD protein were coated in 96 well ELISA plate and different true positive and true negative serum concentrations were screened in checkerboard titrations. The rgD protein was diluted in 0.05 M carbonate-bicarbonate buffer (pH 9.6) to concentrations ranging from 100 to 1.56 ng/well and was tested against varying dilutions (1:20 to 1:20480) of true positive and true negative serum, prepared in PBS-T. The 96-well microtiter plates (Nunc-Immuno Micro Well Poly Sorp, Nunc) were coated overnight at 4°C with purified rgD protein, washed thrice with PBS-T and incubated in blocking solution (5% of skimmed milk powder in PBS-T) for 1h at 37°C. After three washes, serum samples diluted in PBS-T were added in triplicate to the wells and plates were incubated for another 2h at 37°C. After three additional washes, horseradish peroxidase (HRP)-conjugated rabbit anti-bovine immunoglobulin G (IgG; Bethyl Laboratories.Inc) was added at dilution 1:8000 in PBS-T and incubated at 37°C for 1h. Subsequently, the plates were washed thrice with PBS-T and 100 μL/well of peroxidase substrate o-phenylenediamine dihydrochloride (OPD; Sigma Aldrich) diluted in 0.07 mM citrate-phosphate buffer (pH 4.2) with 0.03% (v/v) of H2O2. Plates were incubated for 15 minutes in the dark and the reaction was stopped with 2N H2SO4. Results were read using ELISA Microplate Reader (BIORAD Model 680) at an optical density of 492 nm. The antigen and serum dilutions that produced the maximum difference in absorbance at 492nm between positive and negative (P/N) were selected as strong positive and negative for further standardization. Taking five true negative serum samples, a cut-off value for the recombinant antigen based indirect ELISA was calculated.
Field serum samples were collected from local slaughter house (Bareilly, U.P.) and from Bihar Veterinary College, Patna (twenty-six samples and sixty-nine samples respectively) and stored at -20°C prior to use in the diagnostic procedures. Positive and negative samples to BoHV-1 (as defined by commercially available kit) were used as controls.
Calculation of the cut-off point
The cut-off point was calculated by the mean OD of the negative sera, and three standard deviations (cut-off point= mean OD + 3 standard deviations) (Classen et al., 1987; Frederic et al., 2016).
RESULTS
Amplification and construction of expression cassette
The immunogenic region of gD gene was amplified by PCR using gene specific primers, which resulted in 834bp product (Figure 1). The amplified product was cloned into pRhamTM N-His SUMO Kan vector and expressed in E.cloni cells. The presence of the insert in the recombinant colonies was confirmed by colony PCR (Figure 2). The positive colonies were grown and plasmid DNA was purified and confirmed by sequencing which showed 99% and100% identity of sequences of gD gene of BoHV-1 corresponding to complete genome sequence and Indian isolate respectively in NCBI database (AJ004801.1 and EU523745.1) (Supplementary Figures S1 and S2).
Expression and purification of recombinant gD
PCR positive recombinant clone was selected and induced to express rgD with Rhamnose. On analysis of the expressed protein in SDS-PAGE, a band of ~45 kDa was observed as per the expectation. The expression conditions were optimized to 8h of induction with 0.15% Rhamnose at 32°C (Supplementary Figure S3). The protein was expressed in insoluble form as inclusion bodies, purified under denaturation condition by Ni-NTA purification method followed by refolding with decreasing concentration of urea and the purified expressed protein was obtained. The elution buffer was optimized with 400mM imidazole for maximum yield (Figure 3). The concentration of purified rBoHV-1gD was 3mg/ml from 100ml of culture. Dialyzed protein was finally confirmed by western blot analysis using anti Ni- HRP Probe (Figure 4).
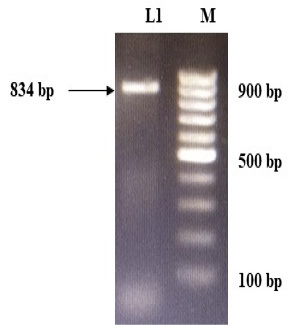
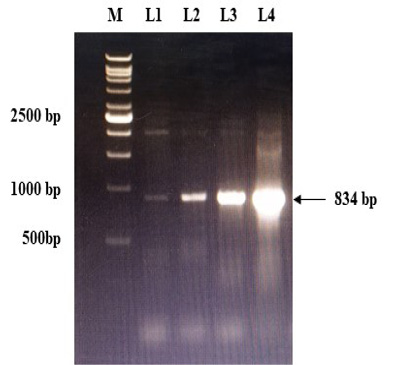
Figure 2: Confirmation of recombinant plasmids (gD) by PCR. L1 to L4: Positive recombinant plasmids showing PCR product of 834 bp; M: VC1Kb DNA Ladder.

Figure 3: A 12% SDS-PAGE of purified rgD protine eluted in different fraction using Ni-NTA affinity chromatography column. L1: Sample flow through; L2: Eluate with wash buffer; M: Magspin-29 protein ladder, 10-180kDa; L3 to L9: Different fraction of eluates.

Figure 4: Confirmation of dialysed rgD protein by western blot using Ni-HRP Probe. L1: Dialysed rgD protein (45kDa); M: Protein Ladder 10-250kDa.
Recombinant gD protein (rgD) based indirect ELISA
The immunogenicity of expressed protein was assessed by BoHV-1 specific antibodies of bovine sera which showed reactivity with rgD in ELISA. Optimum antigen (12.5ng) and sera (1:320) concentration for each well of the plate were determined by checkerboard titrations with conjugate dilution of 1:8000. The P/N value was calculated and the results were interpreted graphically (Figure 5). The cut-off value was calculated (0.238) by the mean OD of the negative sera and three standard deviations (cut-off point = mean OD + 3 standard deviations). The optimum reactivity of the rgD was assessed with known positive and negative sera from commercially available kit (PrioCHECK™ Bovine IBR gB Ab Serum Plate Kit, Applied Biosystems). The relative sensitivity and specificity of the assay were 69% and 52.3% respectively. A few virus-positive and negative sera (confirmed by this study ELISA method and commercial kit) were also tested by western blot analysis (Figure 6) for confirmation of immunoreactivity of rgD for its diagnostic potential.
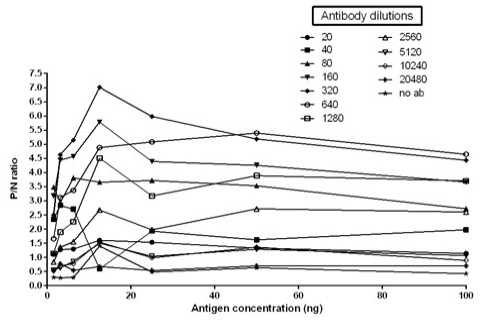
Figure 5: Graph illustrating optimal antigen concentration and antibody dilution used for calculation of cut off value. Optimal antigen concentration: 12.5ng; Optimal antibody dilution: 1:320

Figure 6: Western bolt analysis of expressed rgD protein with bovine field sera: a and b: negative field serum; c and d: Positive field serum giving band of rgD (45 kDa); M: Protein Laddwr 10-250kDa.
DISCUSSION
Bovine herpesvirus 1 is responsible for respiratory disease, abortion and decrease in milk production mainly in cattle and buffalo. It is an important cause of shipping fever and causes huge economic loss to the cattle industry (Barkha et al., 2014). Glycoprotein D is one of the important structural proteins of BoHV1. It has been proposed as the principal vaccine candidate because it induces a consistent and stronger cellular immune response than the others and antibodies against gD have the highest neutralizing titers (Dubuisson et al., 1992; Hughes et al., 1988). Molecular characterization of this gene has provided a good foundation for further studies on this particular gene to be used as a potential candidate for diagnostics.
The partial but immunogenic gD gene sequence was amplified by PCR, cloned into pRhamTM N-His SUMO Kan vector. The expression of rgD was followed with induction by using 0.15% Rhamnose at 32°C. A 45kDa of protein band was seen in case of induced culture as early at four h post induction (pi). The expression of the protein increased at a maximum of up to eight h pi. The proteins resolved in SDS-PAGE were transferred on to a nitrocellulose membrane and detected by BoHV-1 specific serum or Ni-HRP Probe. On analysis, ~45kDa molecular size was observed and confirmed that the expressed recombinant protein was specific to BoHV-1. The predicted size of the expressed protein was 32kDa while fusion His-tag contributed another 13kDa. To assess the antigenic potential of the expressed protein, Ni-NTA affinity columns were used for purification. This method has successfully been used for the purification of several other expressed proteins (Yu et al., 2006; Sun et al., 2007; Yadav et al., 2009). Though, various concentrations of imidazole (100–500mM) have been reported in purifying expressed protein (Yu et al., 2006; Latha et al., 2007; Pathak et al., 2008; Yadav et al., 2009), we found400mM imidazole concentration optimum for elution of protein through affinity column. Purification of protein was followed under denaturing conditions in decreasing urea concentration, which resulted in a good yield of protein in native form and reacted well in the confirmatory assays. Renaturation helps to retain the immunogenicity of recombinant protein, and this has been evidenced previously (Finzi et al., 2003). It is well known that the prokaryotic expression system is cost-effective, user-friendly and easy to scale up commercially. In many of ELISA tests, a prokaryotic system based recombinant protein have been used (Balamurugan et al., 2010). ELISA is one of the most sensitive and extensively applied methods to evaluate expressed recombinant protein (Choi et al., 2005b; Li et al., 2006), where the expressed protein was tested for its suitability as diagnostic antigen in indirect ELISA. Recombinant gD protein used in the study, ELISA reacted well with the antibodies against BoHV-1 and therefore making it evident that the epitopes present in expressed protein are well recognized by the serum antibodies. In order to determine the relative specificity and sensitivity of the assay, known positive and known negative sera with respect to BoHV-1 antibodies were selected on the basis of preliminary screening by a commercially available kit (PrioCHECK™ Bovine IBR gB Ab Serum Plate Kit, Applied Biosystems). The cut-off value is very important factors in a serodiagnostic test above which animals are classified as positive and below which they are considered negative. The accuracy of the cut-off has considerable herd health and economic implications. This is the basis for calculation of diagnostic sensitivity and specificity. Therefore, it must be selected with utmost care. A cut-off value of 0.238 was determined by the mean OD of five negative sera, and to that three-fold standard deviation was added. The relative sensitivity and specificity of the assay were 69% and 52.3% respectively. Sensitivity and specificity in ELISAs can vary widely by type of ELISA (competitive, indirect, or sandwich ELISA), antigens, and mAbs employed, detection system using chemiluminescence or fluorescence, and will have to be determined experimentally. This is an indirect ELISA where immunogenic recombinant protein expressed in prokaryotic system is coated in order to detect IBR positive serum samples. Low sensitivity and specificity of this test may be because of low number of serum samples tested and most of the tests available in market are based on blocking ELISA where monoclonal antibody is used to increase its sensitivity. Since many enzyme immunoassays for the serological diagnosis of BoHV-1 are being marketed around the world however, they are not cost effective in India’s perspective. Hence, this in-house developed indirect ELISA can be economic and user-friendly test for screening a large number of BoHV1 infected bovine population giving an insight towards prevalence of this disease so that preventive measures can be taken to isolate healthy animals from infected population.
ACKNOWLEDGEMENTS
We acknowledge the Department of Biotechnology (DBT) New Delhi, India for providing the funds to carry out this study. We also acknowledge the Director, Indian Veterinary Research Institute, Izatnagar, Bareilly, India for providing the facilities related to this work. We also acknowledge local slaughterhouse (Bareilly, UP) and Bihar Veterinary College, Patna, for providing serum samples.
Authors Contributions
BR and AK designed the experiment. BR, SP and PM performed all experiments. MS contributed reagents/materials and NKC helped in editing the manuscript. All authors reviewed and critically revised the manuscript.
Conflict of Interest
The authors declare that they have no conflicts of interest.
REFERENCES

Supplementary Figure S1: Sequencing of recombinant gD gave 100% identity with partial cds of BoHV-1 in NCBI database accession number: EU523745.1

Supplementary Figure S2: Sequencing of recombinant gD gave 99% identity with complete genome of BoHV-1 in NCBI database accession number: AJ004801.1
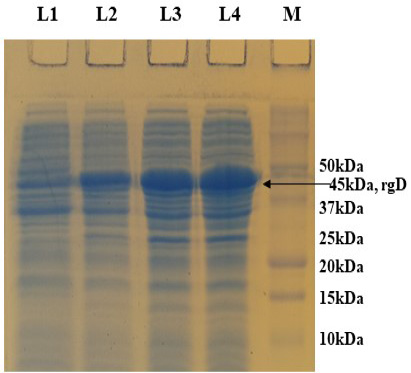
Supplementary Figure S3: 12% SDS-PAGE showing rgD protein expressed at 32 oC induced with 0.15% Rhamnose, collected at different time intervals. L1: Uninduced; L2: 4 hr post induction; L3: 6 hr post induction; L4: 8 hr post induction; M: Protein Ladder (Precision Plus, BIO-RAD).

